499 - Risk for ESKD Progression among Children with Cystinosis: A NAPRTCS Study
Saturday, April 25, 2026
3:30pm - 5:45pm ET
Publication Number: 2485.499
Reeti Kumar, Duke University School of Medicine, Durham, NC, United States; Bradley A. Warady, Children's Mercy Kansas City, Kansas City, MO, United States; Joshua Zaritsky, Phoenix children's hospital, Phoenix, AZ, United States; Laurel Willig, Children's Mercy Kansas City, Kansas City, MO, United States; Larry A. Greenbaum, Children's Healthcare of Atlanta, Decatur, GA, United States; Ewa Elenberg, Baylor College of Medicine, Houston, TX, United States; Daryl Okamura, Seattle Children's, Seattle, WA, United States; Paul Grimm, Stanford University School of Medicine, Stanford, CA, United States; Jodi Smith, Seattle Children's, Seattle, WA, United States
Assistant Professor Duke University School of Medicine Duke University Durham, North Carolina, United States
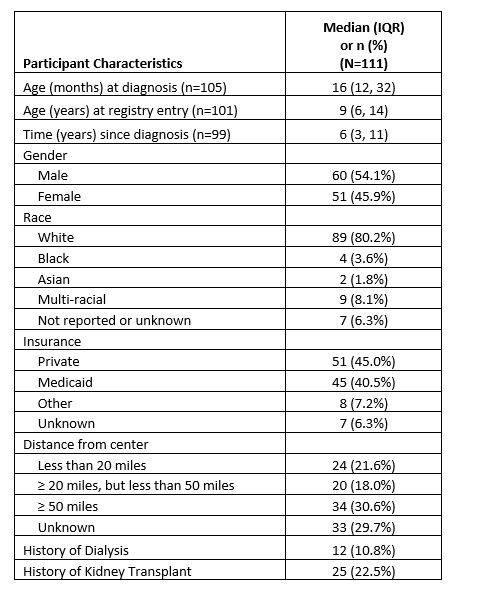
Background: The North American Pediatric Renal Trials & Collaborative Studies (NAPRTCS) Cystinosis Registry is a prospective cohort study of children & young adults with cystinosis. Objective: To describe longitudinal outcomes of patients enrolled in the NAPRTCS Cystinosis Registry. Design/Methods: Children & young adults ( < 25 yrs) with cystinosis at any NAPRTCS center are eligible for enrollment. We summarized demographic and clinical data collected at time of initial presentation, at time of registry entry and every 6 months thereafter. Time to ESKD, defined as either glomerular filtration rate (GFR) < 15ml/min/1.73m2, initiating dialysis or having received a kidney transplant, was calculated from date of diagnosis and displayed with Kaplan-Meier curves. Hazard ratios were calculated with univariable Cox proportional hazard models. Results: Data were collected from 33 centers on 111 subjects who were diagnosed with cystinosis from 12/1999 to 12/2024. The median age at diagnosis was 16 months, with 54.1% male and 80.2% white. 40.5% of subjects were insured by Medicaid and 30.6% of subject traveled 50 miles or more to receive care for cystinosis (Table 1). The median duration of clinical findings prior to diagnosis was 5 months. 50% of the cohort reached end stage kidney disease (ESKD) by 12 years post-diagnosis (Figure 1). The risk of progressing to ESKD within 10 years among subjects who were 'sometimes' prescribed a cystine depleting agent was 1.7 (95% CI 0.6,4.6) times higher compared with subjects who were 'always' prescribed a cystine depleting agent (Figure 2). After initiating cystine depleting agents, subjects who had one WBC cystine level above the recommended threshold (mean WBC cystine concentration of >1.9 nmol ½ cystine/mg protein for Granulocyte cystine assay or >1 nmol ½ cystine/mg protein for Leukocyte cystine assay) had a 13.0 (4.0, 42.1) times higher risk of ESKD relative to someone who never had a high WBC cystine value.
Conclusion(s): Children with cystinosis present early in life with majority of patients diagnosed under 3 years of age. There is a 50% risk of progressing to ESKD within 12 years of diagnosis. Consistent prescription and adherence to cystine depleting agents is essential for preventing progression of kidney disease.
Table 1. Demographics
Figure 1. Overall time to ESKD from cystinosis diagnosis
Figure 2: Time to ESKD from cystinosis diagnosis by receipt of cysteamine bitartrate

.png)
.png)
